TURBOMOLE
TURBOMOLE is a fast and robust quantum chemistry program package with very efficient implementations of various computational methods (HF/DFT/MP2/CC). Properties both for ground and excited states can be obtained. TURBOMOLE has been designed for efficient study of large systems.
Available
- Puhti: 7.5.1, 7.6, 7.7, 7.8
- Mahti: 7.5.1, 7.6, 7.7, 7.8
- Roihu-CPU: 8.0
License
- You may use the Software exclusively for non-profit research purposes.
- Only users from academic (i.e. degree-granting) institutes are allowed to use the Software.
Usage
Initialise TURBOMOLE environment:
On Roihu, jobs must be submitted from the CPU login node (roihu-cpu).
The module sets I_MPI_FABRICS=shm:ofi and MPI_USESRUN=1 automatically.
No manual PATH or MPI setup is needed beyond what is shown in the batch
scripts below. For a full list of available partitions see the
Roihu batch job partitions
page.
Batch scripts
#!/bin/bash
#SBATCH --partition=test
#SBATCH --nodes=2
#SBATCH --ntasks-per-node=40 # MPI tasks per node
#SBATCH --account=<project> # insert here the project to be billed
#SBATCH --time=00:10:00 # time as hh:mm:ss
export PARA_ARCH=MPI
module load turbomole/7.8
export SLURM_CPU_BIND=none
export TURBOTMPDIR=`echo $PWD |cut -d'/' -f1-3`"/TM_TMPDIR/"$SLURM_JOB_ID
mkdir -p $TURBOTMPDIR
export PARNODES=$SLURM_NTASKS
export PATH=$TURBODIR/bin/`$TURBODIR/scripts/sysname`:$PATH
jobex -ri -c 300 > jobex.out
#!/bin/bash
#SBATCH --partition=test
#SBATCH --nodes=1
#SBATCH --cpus-per-task=40 # SMP threads
#SBATCH --account=<project> # insert here the project to be billed
#SBATCH --time=00:10:00 # time as hh:mm:ss
export PARA_ARCH=SMP
module load turbomole/7.8
export TURBOTMPDIR=`echo $PWD |cut -d'/' -f1-3`"/TM_TMPDIR/"$SLURM_JOB_ID
mkdir -p $TURBOTMPDIR
export PARNODES=$SLURM_CPUS_PER_TASK
export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK
export PATH=$TURBODIR/bin/`$TURBODIR/scripts/sysname`:$PATH
jobex -ri -c 300 > jobex.out
#!/bin/bash
#SBATCH --partition=small
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=40 # MPI tasks per node
#SBATCH --account=<project> # insert here the project to be billed
#SBATCH --time=00:10:00 # time as hh:mm:ss
#SBATCH --gres=nvme:100 # requested local disk in GB
export PARA_ARCH=MPI
module load turbomole/7.8
export SLURM_CPU_BIND=none
export TURBOTMPDIR=$LOCAL_SCRATCH/$SLURM_JOBID
mkdir -p $TURBOTMPDIR
export PARNODES=$SLURM_NTASKS
export PATH=$TURBODIR/bin/`$TURBODIR/scripts/sysname`:$PATH
dscf > dscf.out
ccsdf12 > ccsdt.out
#!/bin/bash
#SBATCH --partition=medium
#SBATCH --nodes=2
#SBATCH --ntasks-per-node=128 # MPI tasks per node
#SBATCH --account=<project> # insert here the project to be billed
#SBATCH --time=00:60:00 # time as hh:mm:ss
export PARA_ARCH=MPI
module load turbomole/7.8
export SLURM_CPU_BIND=none
export TURBOTMPDIR=`echo $PWD |cut -d'/' -f1-3`"/TM_TMPDIR/"$SLURM_JOB_ID
mkdir -p $TURBOTMPDIR
export PARNODES=$SLURM_NTASKS
export PATH=$TURBODIR/bin/`$TURBODIR/scripts/sysname`:$PATH
jobex -ri -c 300 > jobex.out
On Roihu the module sets MPI_USESRUN=1 so TURBOMOLE launches tasks via
srun automatically. No wrapper script is needed.
Note
Local NVMe disk is not yet available for standard M-node jobs on Roihu. Scratch I/O goes to Lustre. NVMe support will be enabled in a future update.
#!/bin/bash
#SBATCH --partition=small # see batch-job-partitions for all options
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=12 # MPI tasks per node
#SBATCH --mem-per-cpu=2000 # MB per CPU core
#SBATCH --account=<project> # insert here the project to be billed
#SBATCH --time=01:00:00 # time as hh:mm:ss
export PARA_ARCH=MPI
module load turbomole/8.0
export SLURM_CPU_BIND=none
export PATH=$TURBODIR/bin/$(sysname):$PATH
export TURBOTMPDIR=/scratch/<project>/<user>/TM_TMPDIR/$SLURM_JOB_ID
mkdir -p $TURBOTMPDIR
export PARNODES=$SLURM_NTASKS
jobex -ri -c 300 > jobex.out
#!/bin/bash
#SBATCH --partition=small # see batch-job-partitions for all options
#SBATCH --nodes=1
#SBATCH --cpus-per-task=12 # SMP threads
#SBATCH --mem-per-cpu=2000 # MB per CPU core
#SBATCH --account=<project> # insert here the project to be billed
#SBATCH --time=01:00:00 # time as hh:mm:ss
export PARA_ARCH=SMP
module load turbomole/8.0
export PATH=$TURBODIR/bin/$(sysname):$PATH
export TURBOTMPDIR=/scratch/<project>/<user>/TM_TMPDIR/$SLURM_JOB_ID
mkdir -p $TURBOTMPDIR
export PARNODES=$SLURM_CPUS_PER_TASK
export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK
jobex -ri -c 300 > jobex.out
Note
Occasionally mpshift calculations are terminated due to the local /tmp
becoming full. The problem can be circumvented by redefining $TMPDIR:
Note
Particularly some of the wavefunction-based electron correlation methods can be very disk I/O intensive. Such jobs benefit from using the fast local storage on Puhti. Using local disk for such jobs will also reduce the load on the Lustre parallel file system. On Roihu, local NVMe is not yet available for standard M-node jobs.
Performance example
Here we provide a brief example of how different resource allocations affect TURBOMOLE's performance. We use α-Tocopherol (vitamin E, C29H50O2, 81 atoms) as the input structure. The geometry is available at vite_wb97x_d4_roihu.sh.
The tests were conducted in a production environment where job interference may introduce performance fluctuations.
The following figure shows the wall time for a ωB97X-D4/def2-TZVP single-point energy calculation as a function of the number of cores/threads on Roihu, using both SMP (OpenMP) and MPI parallelism.
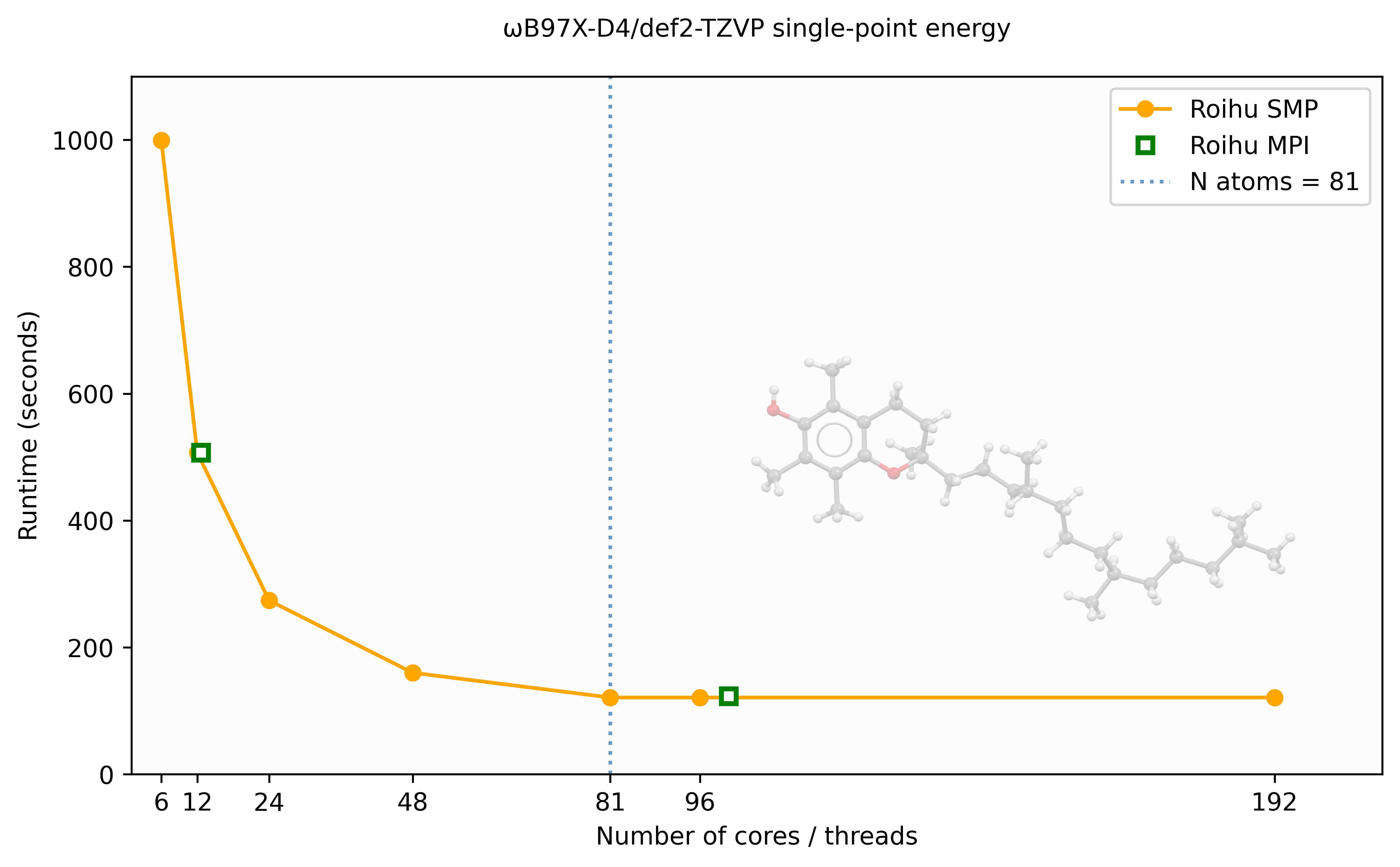
SMP (PARA_ARCH=SMP) scales well up to approximately 48 threads for this
81-atom system, after which performance levels off. The MPI (PARA_ARCH=MPI)
results show similar wall times to SMP but require significantly more memory
— approximately 15× more at 96 processes for this system.
For ridft DFT calculations on a single node, SMP is therefore generally
preferred over MPI. The scaling limit is related to the number of atoms —
larger molecules will benefit from more threads before reaching saturation.
After the job completes, check actual memory and wall time usage with:
NumForce calculations
NumForce is a tool that can be used to calculate second derivatives (molecular
Hessian) for all methods for which analytic gradients are available in
TURBOMOLE. A NumForce job spawns 3*N*2 (N = number of atoms) independent
gradient calculations. Usually it is most efficient that the single gradient
calculations are run as serial (unset PARA_ARCH). Each serial calculation is
expected to take roughly the same time, hence optimally the total number of
gradient calculations should be an integer multiple of the allocated cores.
A NumForce step in a job file:
unset PARA_ARCH
export HOSTS_FILE=$PWD/turbomole.machines
rm -f $HOSTS_FILE
srun hostname > $HOSTS_FILE
ulimit -s unlimited
kdg tmpdir
NumForce -ri -central -mfile $HOSTS_FILE > NumForce.out
References
Please quote the usage of the program package under consideration of the version number:
- TURBOMOLE V8.0, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from https://www.turbomole.org
- A review article should be mentioned, as well: https://doi.org/10.1063/5.0004635
- Scientific publications require proper citation of methods and procedures employed. The output headers of TURBOMOLE modules include the relevant papers.